A head-to-head benchmark exhibits why the latest algorithm will not be all the time your best option and the way sequencing context can decide which methylation caller researchers ought to belief.

Research: Complete benchmarking of instruments for nanopore-based detection of DNA methylation. Picture Credit score: AI-generated utilizing ChatGPT/OpenAI
Nanopore sequencing can detect DNA base modifications instantly from native DNA, however precisely figuring out these chemical marks stays difficult. A current paper, revealed on-line as an ‘Article in Press’ in Nature Communications, systematically benchmarked software program instruments for detecting DNA modifications from nanopore sequencing knowledge.
Researchers discovered that particular newer fashions provided one of the best total efficiency for non-CpG 5-methylcytosine (5mC), which means methylated cytosines exterior cytosine-guanine, or CpG, websites, 6-methyladenine (6mA), and 4-methylcytosine (4mC), whereas the older Dorado v4r1 mannequin and RockFish remained essentially the most dependable for CpG methylation profiling. These findings present sensible steerage for choosing computational instruments for epigenetic analysis and nanopore-based genomic evaluation.
The Position of Nanopore Sequencing
DNA methylation is a vital epigenetic modification that contributes to the regulation of gene expression, genome stability, and mobile growth. Conventional strategies for detecting these modifications, resembling bisulfite sequencing, require chemical remedy that may harm DNA, introduce amplification bias, and complicate mapping in complicated genomic areas.
On this context, nanopore sequencing supplies a direct different by analyzing native DNA with out chemical conversion. As particular person DNA molecules go by a bioengineered nanopore, adjustments in electrical present reveal the presence of modified bases.
Oxford Nanopore’s R10 stream cells use a extra steady, bioengineered nanopore with an extended sensing area, enhancing the decision of homopolymer sequences and total sequencing accuracy. Neural network-based algorithms then interpret these electrical indicators to determine DNA base modifications alongside the nucleotide sequence. As sequencing accuracy has improved, the principle problem has shifted from sign acquisition to the computational interpretation of those complicated electrical indicators.
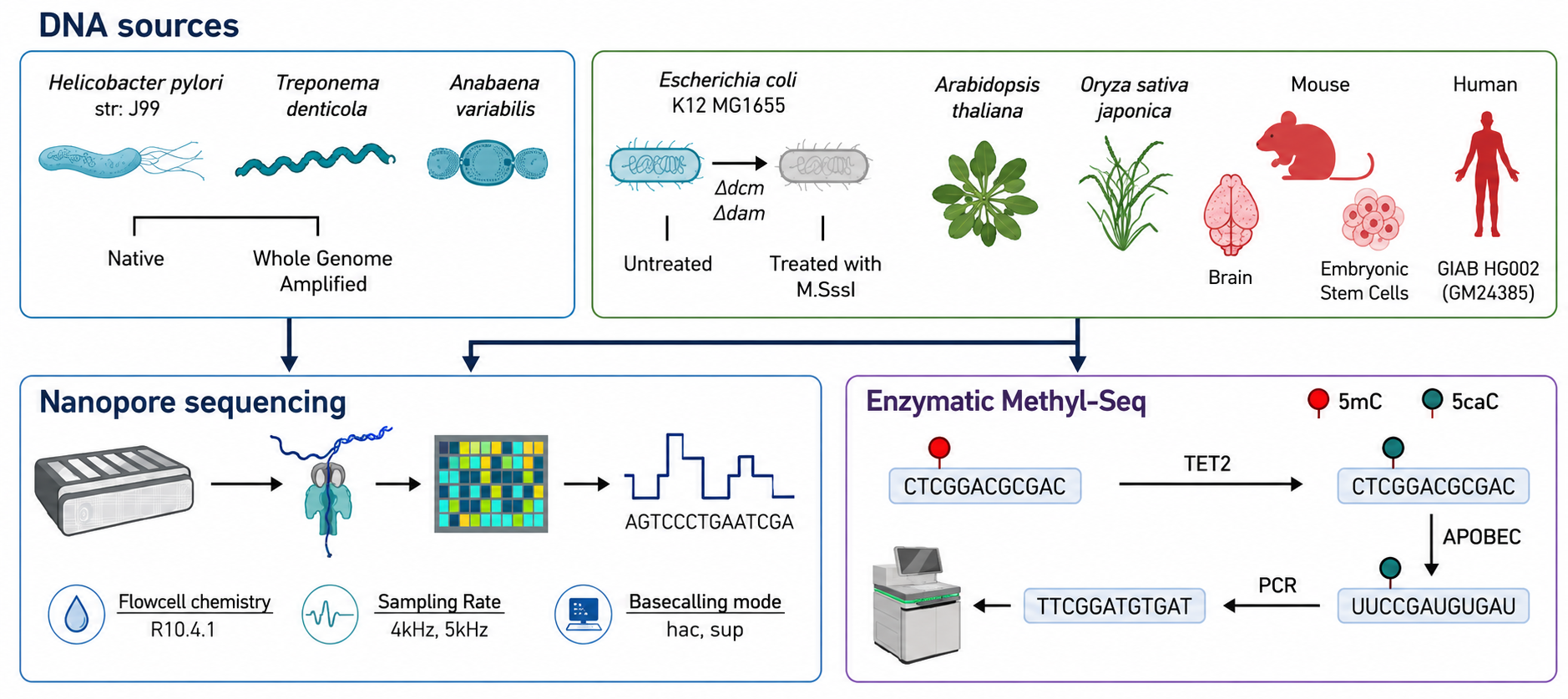
Sources of DNA and complete genome sequencing workflow used on this research. Genomic DNA from bacterial/plant/mammalian samples was sequenced on the R10.4.1 flowcells. A subset of samples (proper field on the highest) had been additionally topic to Enzymatic Methyl-Seq (EMSeq) as floor reality for 5-methylcytosine (5mC). Picture tailored from fig 1a. Kulkarni, O., et al. (2026). Complete benchmarking of instruments for nanopore-based detection of DNA methylation. Nat Commun. DOI: 10.1038/s41467-026-75183-6 utilizing ChatGPT/OpenAI.
Framework for Benchmarking Methylation Instruments
To guage present computational strategies for nanopore methylation evaluation, researchers benchmarked broadly used software program utilizing whole-genome sequencing knowledge from numerous organic sources. These knowledge had been generated from 5 bacterial species, two plant species, and mammalian samples, together with mouse whole-brain tissue, mouse embryonic stem cells, and the human HG002 cell line. This numerous dataset captured a broad vary of DNA modifications, from the frequent methylation patterns present in mammals to much less frequent modifications current in vegetation and micro organism.
The datasets included sequencing knowledge generated utilizing Oxford Nanopore R10.4.1 stream cells at sampling charges of 4 kHz and 5 kHz. Enzymatic Methyl-seq supplied the 5mC reference knowledge for the plant and mouse samples and E. coli, whereas the human HG002 knowledge had been in contrast with a publicly accessible whole-genome bisulfite sequencing dataset. Public Pacific Biosciences knowledge supplied 6mA and 4mC comparisons for 3 bacterial species, whereas REBASE motif data was used for the opposite two species.
The uncooked nanopore indicators had been processed with DeepBAM, DeepMod2, DeepPlant, f5C, RockFish, and a number of variations of the Dorado fashions. The benchmark in contrast every mannequin underneath completely different working modes, together with high-accuracy and super-accuracy settings. The research additionally evaluated detection accuracy, false-positive charges, processing velocity, reminiscence use, and the results of sequencing depth, learn high quality, and neighboring DNA modifications by evaluating nanopore predictions with the reference datasets.
Efficiency and Algorithmic Limitations
The benchmark confirmed clear variations in efficiency throughout computational fashions. For normal CpG methylation, Dorado v4r1 and RockFish achieved the very best accuracy and settlement with the reference datasets. Though newer Dorado fashions confirmed decrease accuracy in routine CpG methylation profiling as a consequence of increased false-negative charges, Dorado v5r3 carried out finest total for non-CpG 5mC and 4mC, whereas Dorado v5r1 was most well-liked total for 6mA. The most recent v5.2 fashions diminished some false-positive results from neighboring modifications however weren’t persistently superior as a result of a number of had poorer recall.
The evaluation additionally recognized vital limitations shared by many algorithms. As a result of {the electrical} sign measured by a nanopore displays a number of neighboring bases, close by DNA modifications may enhance false-positive or false-negative calls relying on the modification, sequence context, distance, and mannequin.
DeepPlant carried out nicely for non-CpG methylation in plant knowledge when particular person DNA reads had been evaluated, however carried out poorly on mammalian datasets, indicating a powerful species-specific coaching bias. Dorado v5r3 usually supplied higher efficiency when methylation estimates had been aggregated at particular person genomic websites. Computational efficiency additionally assorted significantly. Dorado supplied the very best total throughput within the bacterial benchmarks whereas utilizing 12–14 GB of reminiscence, whereas DeepPlant required a median of greater than 84 GB. Nonetheless, f5C outperformed the super-accuracy Dorado 5mCG mannequin in each velocity and reminiscence use.
The research additionally discovered that correlation in rice CpG analyses usually plateaued after a median protection of about 20×, though error continued to lower at increased protection. The authors subsequently beneficial no less than 20× median protection. In three bacterial 6mA datasets, filtering reads beneath a Phred high quality rating of 20 diminished quality-dependent undercalling and produced extra constant methylation estimates.
Research Limitations
The research used Enzymatic Methyl-seq as reference knowledge regardless of its potential biases, didn’t benchmark 5-hydroxymethylcytosine, and evaluated 4mC throughout comparatively few sequence contexts. It additionally relied on REBASE somewhat than on direct Pacific Biosciences knowledge for 2 bacterial species, and a few analyses had been restricted to chromosome 1 or required massive datasets to be cut up into smaller computational batches.
Implications for Epigenetic Analysis
This analysis affords sensible steerage for choosing computational instruments for nanopore-based epigenetic evaluation. By matching algorithms to particular DNA modifications, scientists can enhance the accuracy of methylation profiling whereas decreasing analytical errors. These findings are notably beneficial for plant genomics, the place correct detection of non-CpG methylation can help analysis into growth, stress responses, and transposon silencing.
Extra usually, dependable detection of DNA methylation from native DNA helps the usage of nanopore sequencing in epigenetic research. Nonetheless, this benchmarking research didn’t consider scientific samples, diagnostic accuracy, precision medication purposes, crop traits, or illness biomarkers. As computational strategies advance, nanopore sequencing could change into a extra reliable device for finding out disease-associated epigenetic adjustments and different related biomarkers, however these potential purposes require separate validation.
Future Instructions in Nanopore Sequencing
In abstract, this benchmarking research supplies a complete evaluation of present nanopore methylation evaluation instruments, highlighting each their strengths and their limitations. Whereas advances in nanopore sequencing {hardware} have improved knowledge high quality, correct interpretation of DNA modifications nonetheless depends upon strong computational strategies.
Future work ought to give attention to additional creating algorithms that higher account for the affect of neighboring DNA modifications whereas sustaining excessive accuracy and computational effectivity. The open-access datasets and benchmarking framework established by the researchers present a beneficial useful resource for refining nanopore methylation evaluation and supporting future advances in epigenetics and genomics.